GATK教程 / 变异检测前的数据预处理
前文回顾1. GATK官方教程 / 概述及工作前的布置2. GATK教程 / 体细胞短变异检测 (SNV+InDel)流程概览Data pre-processing for variant discovery目的 这是第一阶段的工作,必须在所有变异发现之前进行。它涉及对原始序列数据(以FASTQ或uBAM格式提供)进行预处理,以产生可供分析的BAM文件。 这涉及到对参...
前文回顾
2. GATK教程 / 体细胞短变异检测 (SNV+InDel)流程概览
Data pre-processing for variant discovery
目的
这是第一阶段的工作,必须在所有变异发现之前进行。它涉及对原始序列数据(以FASTQ或uBAM格式提供)进行预处理,以产生可供分析的BAM文件。
这涉及到对参考基因组的比对,以及一些数据清理操作,以纠正技术偏差,使数据适合分析。
参考实现
|
管道 |
结果 |
笔记 |
Github |
Terra |
|
Prod* 胚系短变体每单样本Calling |
uBAM到GVCF |
GCP优化 |
有 |
hg38 & b37 |
|
Genome Analysis Pipeline |
uBAM到GVCF或队列/cohort VCF |
GCP优化 |
有 |
hg38 |
|
通用的数据预处理 |
uBAM到分析就绪的BAM |
通用 |
有 |
hg38 & b37 |
* Prod:指的是Broad研究所的数据科学平台生产管道 (Broad Institute's Data Sciences Platform production pipelines),用于处理Broad的基因组测序平台设备产生的序列数据。
预期的输入文件
这个工作流旨在对单个样本进行操作,对于这些样本,数据最初被组织在称为读组 (Read groups)的不同子集中。这些相当于文库(即从生物样本中提取、并准备进行测序的DNA产物,包括使用识别条形码进行片段化和标记)与测序通道(即Lane:DNA测序芯片上的物理分隔单元)的“交叉”。此“交叉”是由于多路复用(即Multiplexing:混合多个库、并在多个通道上进行测序的过程,以降低风险和人为操作所带来的影响)所产生的。
我们的参考实践流程,期望测序数据以未比对的BAM (Unmapped BAM, uBAM)文件格式输入。一些转换程序可将FASTQ转换为uBAM文件。
主要步骤
① 首先将读取的序列映射到参考基因组,生成一个按坐标排序的SAM/BAM格式的文件。即全基因组比对。
② 接下来,对重复序列进行标记,以减轻一些数据生成步骤(如PCR扩增)中带来的偏差。
③ 最后,重新校准基本质量分数,因为变异调用算法(Variant calling algorithms)在很大程度上依赖于每个读序列(Sequence Read)取中分配给单个碱基调用的质量分数(Quality scores)。
映射到参考基因组
涉及的工具:BWA, MergeBamAlignments
http://bio-bwa.sourceforge.net/
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#MergeBamAlignment
第一个处理步骤是逐读组(Per-read group)执行的,包括将每个读对(Read pair)映射到参考基因组。
参考基因组由公共的基因组序列所合并成的单链表示(如人类hg38基因组),旨在为所有基因组分析提供一个共同的坐标框架。因为映射/比对算法(Mapping algorithm)是单独处理每个读对(Read pair)的,所以可被大规模并行化,以根据需要来增加数据处理的吞吐量。
标记重复 (Mark Duplicates)
涉及工具:MarkDuplicatesSpark / MarkDuplicates + SortSam
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#MarkDuplicatesSpark
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#MarkDuplicates
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#SortSam
MarkDuplicatesSpark
此第二个处理步骤是对每个样本执行的,包括识别可能来自相同原始DNA片段的副本(Duplicates/重复)的读对,这些“Duplicates/重复”一般是通过一些人工过程(Artifactual processes,如PCR扩增和建库等)。这些被认为是非独立的观察,因此程序在每组重复中标记了除单个读对外的所有读对,导致标记的对(marked pairs)在变异发现过程中默认被忽略。
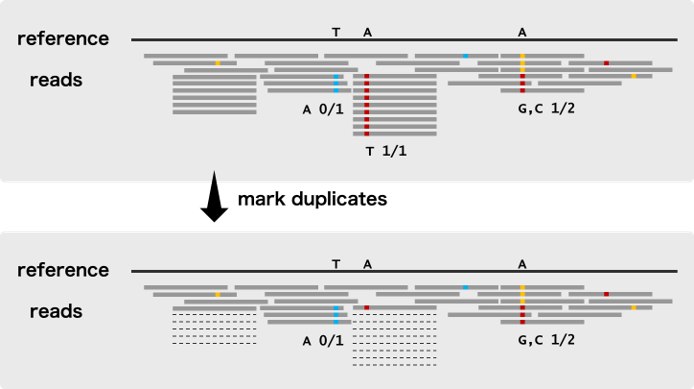
注意上图中的“虚线”Reads,即为“Duplicates”
在测序分析时,duplicate的reads是来源于同一条原始的read,相当于是同一个信息,假如某个位置有100条reads覆盖,90条是duplicate,其实这个位置就相当于90条reads的信息是一个有用信息,如果这个原始read因为测序的问题发生了一个突变,不考虑duplicate的话,就是90个突变,很容易被作为假阳性检出 (90/100),而如果考虑是duplicate,这90个read仅被作为一个信息 (1/10),就不太会被检出了
在这一阶段,读取数据还需要按照坐标顺序进行排序,以便进行下一步的预处理。
MarkDuplicatesSpark执行重复标记步骤和排序步骤,用于此阶段的预处理。由于在一个样本中读取对之间进行大量的比对、比较,这一阶段的“流水线”(即数据处理)一直是一个性能瓶颈,因此MarkDuplicatesSpark利用Apache Spark来并行化进程,以便更好地利用所有可用资源。即使不需要访问专用的Spark集群,这个工具也可以在本地运行。
MarkDuplicates + SortSam
作为MarkDuplicatesSpark的替代方案,这个步骤可以通过使用MarkDuplicates的Picard实现,执行重复序列标记;然后使用SortSam来对Reads进行排序。这两种工具目前都是作为单线程工具实现的,因此不能利用核心并行 (Core parallelism)。
建议在拥有大量核心或能够访问快速磁盘驱动器的设备上,在MarkDuplicatesSpark上运行MarkDuplicates,然后运行SortSam。如果按照最佳实践运行,MarkDuplicatesSpark会产生与这种方法相当的位输出 (Produces bit-wise equivalent output to this approach),因此上述任何一组工具都是有效的。
碱基(质量得分)重新校准/Base (Quality Score) Recalibration
涉及的工具:BaseRecalibrator, Apply Recalibration, AnalyzeCovariates(可选)
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#BaseRecalibrator
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#ApplyBQSR
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#AnalyzeCovariates
这第三个处理步骤是在每个样本中执行的,包括应用机器学习来检测和纠正碱基质量分数(Base quality score)中的系统错误模式(Patterns of systematic errors),其中碱基质量分数是由测序仪对每个碱基给出的置信度分数(Are confidence scores emitted by the sequencer for each base)。
在变异发现过程中,碱基质量分数在权衡支持或反对可能的变异等位基因(Variant alleles)的证据方面,发挥着重要作用,因此纠正数据中观察到的任何系统性偏差是很重要的。
偏差可以来自文库制备和测序过程中的生化过程,或来自芯片的制造缺陷,又或来自测序仪的仪器缺陷(Instrumentation defects in the sequencer)。
重新校准过程包括从数据集中的所有碱基调用(Base calls)中收集协变量测量值(Covariate measurements),从这些统计数据中构建模型,并根据所得到的模型对数据集应用碱基质量调整(Base quality adjustments)。
初始统计数据收集可以通过分散在基因组坐标上进行并行化,通常是通过染色体或染色体批 (Batches)进行并行化,但如果需要,可以进一步分解以提高数据处理的吞吐量。然后每个区域的统计数据必须被收集到一个单独的全基因组范围内的协变模型(A single genome-wide model of covariation)中;这是不能并行化的,但在计算上是微不足道的,因此不是性能瓶颈。
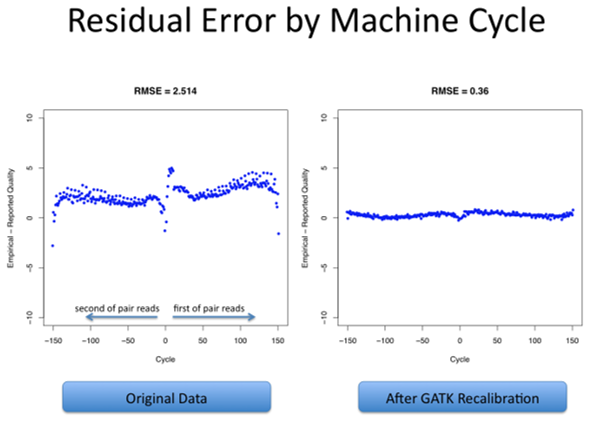
最后,将从模型中导出的重新校准规则(Recalibration rules)应用于原始数据集,以生成重新校准的数据集。这与初始统计数据收集在基因组区域上的并行方式相同,然后进行最终文件合并操作,以生成每个样本的单个分析就绪文件(Analysis-ready file)。
一些问题反馈
需要指出的是,Fastq -> Bam过程中,需要包含读组标签 (Read group tags),因为它们在流程的重新校准碱基质量分数阶段及其之后需要。
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集




魔乐社区(Modelers.cn) 是一个中立、公益的人工智能社区,提供人工智能工具、模型、数据的托管、展示与应用协同服务,为人工智能开发及爱好者搭建开放的学习交流平台。社区通过理事会方式运作,由全产业链共同建设、共同运营、共同享有,推动国产AI生态繁荣发展。
更多推荐

 3
3 0
0- 0



 已为社区贡献63条内容
已为社区贡献63条内容

所有评论(0)