iMeta | 刘永鑫组-开发微生物组数据分析与可视化平台MicrobiomeStatPlots
点击蓝字 关注我们MicrobiomeStatPlots:一个用于微生物组宏组学和生物信息学的统计绘图库iMeta主页:http://www.imeta.science研究论文●原文:iMeta(IF 23.8)●原文链接DOI: https://doi.org/10.1002/imt2.70002●2025年2月17日,中国农业科学院深圳农业基因组研究所刘永鑫等在iMeta在线发表了题...
点击蓝字 关注我们
MicrobiomeStatPlots:一个用于微生物组宏组学和生物信息学的统计绘图库

iMeta主页:http://www.imeta.science
研究论文
● 原文: iMeta (IF 23.8)
● 原文链接DOI: https://doi.org/10.1002/imt2.70002
● 2025年2月17日,中国农业科学院深圳农业基因组研究所刘永鑫等在iMeta在线发表了题为“MicrobiomeStatPlots: Microbiome statistics plotting gallery for meta-omics and bioinformatics”的文章。
● 本研究结合了基本的生物信息学工作流程与多组学流程,同时提供80多种不同的可视化案例,用户不仅可以自定义图表为平台的扩展做出贡献,还可以在 GitHub上免费访问丰富的生物信息学知识。弥补了微生物组数据分析和可视化方面的空白。
● 第一作者:白德凤、马闯、荀佳妮
● 通讯作者:刘永鑫(liuyongxin@caas.cn)、王瑶(wangyao01@caas.cn)
● 合作作者:罗豪、杨海飞、吕虎杰、朱志豪、盖安冉、Salsabeel Yousuf、彭凯、许珊珊、高云云
● 主要单位:中国农业科学院深圳农业基因组研究所、安徽农业大学园艺学院、青岛农业大学生命科学学院、香港理工大学食品科学与营养学系、广东医科大学基础医学院、郑州大学农学院、扬州大学动物医学院、合肥工业大学食品与生物工程学院、北京林业大学生态与自然保护学院
亮 点
● 可视化资源丰富:MicrobiomeStatPlots提供超过80个可重复的可视化案例,并集成多组学分析流程,以促进微生物组数据的分析和解释;
● 可定制且用户友好:该平台包括详细的教程、示例数据集和基于R的绘图代码,使研究人员能够根据自己的需求定制可视化;
● 协作和开源:MicrobiomeStatPlots鼓励社区贡献以扩展其功能,并支持微生物组研究的不断发展。您可以在 https://github.com/YongxinLiu/MicrobiomeStatPlot 上使用它并做出贡献。
摘 要
微生物组研究的快速发展产生了前所未有的多组学数据,对数据分析和可视化提出了挑战。为了解决这些问题,我们推出了微生物组数据分析与可视化(MicrobiomeStatPlots),这是一个综合平台,提供简化、可重复的微生物组数据分析和可视化工具。该平台将基本的生物信息学工作流程与多组学流程相结合,并提供了82种不同的可视化案例来解释微生物组数据集。通过结合基本教程和高级的基于R的可视化策略,MicrobiomeStatPlots提高了研究人员的可访问性和可用性。用户可以自定义图表,为平台的扩展做出贡献,并在GitHub上免费访问丰富的生物信息学知识(https://github.com/YongxinLiu/MicrobiomeStatPlot)。未来计划扩大对代谢组学、病毒组学和宏转录组学的支持,以及将可视化工具无缝集成到组学工作流程中。MicrobiomeStatPlots弥补了微生物组数据分析和可视化方面的空白,为更高效、更有影响力的微生物组研究铺平了道路。
视频解读
Bilibili:https://www.bilibili.com/video/BV1zDAYeZEmD/
Youtube:https://youtu.be/9BGCjsewNAU
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
近年来,组学技术的快速发展彻底改变了微生物群落的表征方式,标志着微生物组研究进入了多组学时代。有多种组学方法用于研究微生物组,包括培养组学、扩增子测序、宏基因组学、宏转录组学、宏蛋白质组学、病毒组学和代谢组学。这些进步推动了功能强大且用户友好的生物信息学工具的开发。扩增子测序分析最常用的工具包括QIIME2和USEARCH。对于宏基因组数据分析,常用的工具包括用于质量控制的Trimmomatic和fastp、用于精确分类的Kraken2、用于全面功能分析的HUMAnN3流程。总之,这些创新为分析和探索各种类型的组学数据提供了重要支持。
近几年,组学数据分析变得越来越容易,用户友好的工具和平台简化了复杂的工作,使用户能够快速适应数据分析任务。例如,STAMP是一款可在本地安装的软件,专为组间差异分析而设计,并提供多种可视化选项。培养组(Culturome)、易扩增子(EasyAmplicon)和易宏基因组(EasyMetagenome)(https://github.com/YongxinLiu/EasyMetagenome) 等流程用于在本地服务器上进行微生物数据分析,分别针对培养组、扩增子和宏基因组分析,并提供可发表的可视化代码。MetaProteomeAnalyzer是一种专门用于分析宏蛋白质组学数据的工作流程,Notame提供了代谢组学分析的工作流程。MicrobiomeAnalyst支持扩增子、宏基因组学和代谢组学数据的分析,而MGV(肠道宏基因组病毒)、ViWrap和Hecatomb等病毒组学工作流程则专用于病毒组数据分析。此外,ImageGP、OmicsStudio、OmicShare工具和Wekemo Bioincloud等综合在线平台提供了丰富多样的在线可视化功能。此外,一些针对特定方面的在线平台和数据库(例如 iNAP、GeNets、IPGA、EVenn和FoodMicroDB)为用户提供了多样化的微生物组数据分析和可视化选项。此外,一些研究开发了常用于组学数据分析的R语言包,而另一些研究则回顾了组学数据分析中常用的R包和软件并提出了最佳的分析实践。从组学数据中识别可靠且稳健的生物标志物的方法也在迅速发展。这些进展表明多组学分析已成为解决科学问题的基石。
尽管取得了这些进展,微生物组数据分析和可视化仍面临挑战。STAMP等工具仅专注于某一类型的差异比较,在覆盖广泛的组学技术和适应该领域的快速迭代方面存在局限性。流程或工作流专注于数据分析,但缺乏数据可视化解释。在线平台有多种可视化选项,但定制有限。它们通常缺乏R代码的可重复性。一些R包可用于微生物组数据分析和绘图,但它们相对分散,需要安装和使用这些包的经验。由于R包经常更新,现有的基于代码的分析可视化工作流程经常缺乏更新,导致一些代码无法运行。全面掌握微生物组数据分析的基本知识和技能通常费时费力。虽然可视化的主要目的是降低数据维度并方便结果解释,但现有的工具和软件往往无法提供足够的解释支持,使用户对结果的洞察力有限。这些限制使研究人员无法有效地分析、可视化和解释他们的数据。
为了应对这些挑战,我们推出了MicrobiomeStatPlots,这是一个用于微生物组数据分析和可视化的综合资源平台。MicrobiomeStatPlots提供了82种基于R语言的可视化样式集合,旨在满足多组学研究人员的不同需求。该资源涵盖培养组学、扩增子测序和宏基因组学,同时还提供生物信息学基础知识的入门教程,例如Shell和Linux的使用、R中的统计分析以及Python中的数据处理和可视化。此外,MicrobiomeStatPlots还为微生物组研究的策略和最佳实践提供了实用指导。MicrobiomeStatPlots通过将基本的生物信息学知识与高级可视化和解释工具相结合而脱颖而出,使其成为新手和经验丰富的研究人员的宝贵平台。该项目可在GitHub上免费访问(https://github.com/YongxinLiu/MicrobiomeStatPlot),鼓励用户贡献并扩展其资源。通过持续更新和新可视化工具的集成,MicrobiomeStatPlots旨在通过为微生物组数据分析提供高效、可重复且用户友好的解决方案,为领域研究提供支持。
结 果
MicrobiomeStatPlots的概述
微生物组研究主要关注微生物、DNA和mRNA。为研究微生物组采用各种组学技术,如培养组学、扩增子测序、宏基因组学、病毒组学、代谢组学、宏转录组学和蛋白质组学(图1A)。MicrobiomeStatPlots通过整合高级数据统计、可视化和结果解释功能,扩展了现有的流程,如培养组(Culturome)、易扩增子(EasyAmplicon)和易宏基因组(EasyMetagenome)。这些改进使研究人员能够有效地分析和理解微生物组数据。该平台还支持开发用于其他组学分析的工作流程(图1B)。MicrobiomeStatPlots提供用于图形可视化的R代码、与图表相关的文献来源的简要介绍以及结果解释,使研究人员更容易识别和实施适当的分析方法(图1C)。MicrobiomeStatPlots支持广泛的科学研究,包括人类疾病和健康、微生物与动物的共生关系以及植物与微生物的相互作用(图1D)。截至2025年1月16日,MicrobiomeStatPlots已获得广泛关注,在GitHub存储库(https://github.com/YongxinLiu/MicrobiomeStatPlot)上有235个forks和399个stars。
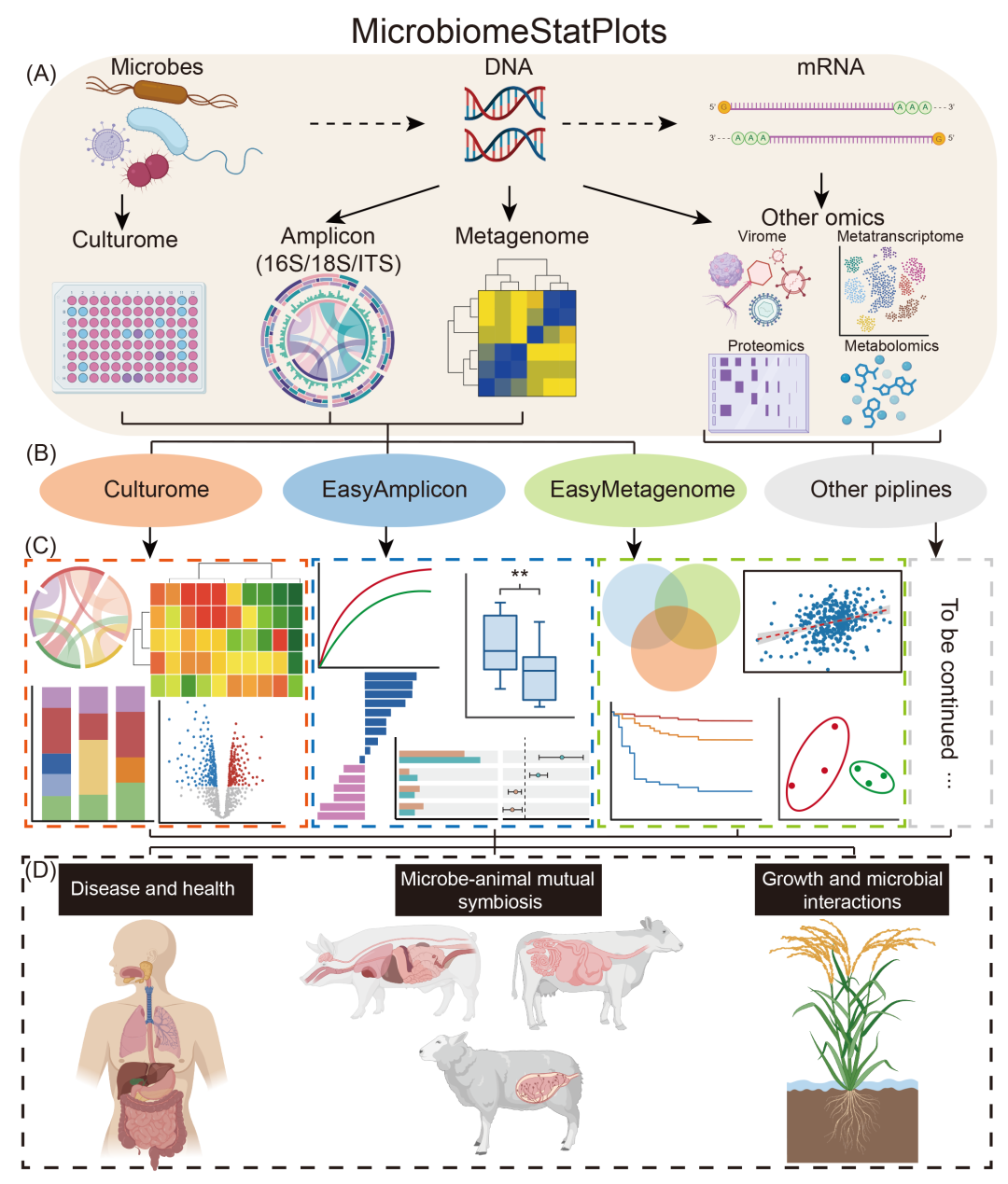
图1. MicrobiomeStatPlots的概述
(A)MicrobiomeStatPlots中分析的多组学数据类型概述。该项目支持各种组学方法,包括培养组、扩增子(16S/18S/ITS)、宏基因组和其他组学(如病毒组、宏转录组、蛋白质组学和代谢组学)。(B)MicrobiomeStatPlots中包含的核心数据分析流程,例如Culturome、EasyAmplicon、EasyMetagenome和其他正在开发的流程。(C)MicrobiomeStatPlots为多组学数据分析生成的示例可视化。这些包括不同数据类型的热图、箱线图、条形图、散点图和维恩图,突出显示它们在了解微生物群落中的应用。(D)MicrobiomeStatPlots在微生物组研究中的应用。这些包括对疾病和健康、微生物与动物共生关系和植物与微生物相互作用的研究,为微生物群落的生态和功能动态提供见解。此图中的某些图形由BioRender(https://app.biorender.com/)创建。
MicrobiomeStatPlots的结构和组成
在培养组学、扩增子测序或宏基因组学中,分析过程都是从原始序列文件开始,经过几个序列处理步骤(图2A)。通过使用已经开发的培养组学、扩增子测序和宏基因组学分析流程,结合第三方软件分析(图2C),生成包含微生物丰度、物种注释和功能特征的特征表格(图2D)。最后,MicrobiomeStatPlots提供的可视化解决方案能够通过R代码生成可重复的图,并将原始数据转换为可解释的图形。该项目提供了微生物组实验设计的方法技巧、使用Shell和Linux的教程、R中的统计分析以及Python中的数据处理和可视化,为研究人员创建了一站式学习平台。它旨在通过持续优化和共享来支持微生物组数据分析、可视化和解释(图2E)。
MicrobiomeStatPlots系统地组织了六个关键领域的微生物组数据分析和解释的知识(图2),包括:(1)用户入门指南,(2)在个人计算机上构建微生物组分析平台以及使用R软件和Linux系统的基本指引,(3)培养组、扩增子和宏基因组的多组学数据分析流程,(4)使用R和Shell脚本可视化和解释微生物组特征表,(5)微生物组数据分析的最佳实践和创新方法,(6)总结其他微生物组数据分析技术的附录。这种结构化方法可确保研究人员可以轻松找到和利用所需的信息。
MicrobiomeStatPlots的可视化和解释部分是更新最频繁的部分,目前包含80多个微生物组数据分析、可视化和解释示例。本节涵盖一系列主题,包括使用基础图(例如散点图、线图和基本条形图)、复合图(例如气泡图、误差线图和曼哈顿图)、alpha或beta多样性可视化(例如PCoA、CPCoA和dbRDA)。此外,它还涵盖了各种微生物组数据分析技术,例如使用MaAsLin2和ANCOMBC进行差异分析、使用anpan(https://huttenhower.sph.harvard.edu/anpan)进行微生物菌株评估以及使用稀疏相关性进行成分数据(SparCC)共丰度网络分析。MicrobiomeStatPlots还包括用于识别微生物生物标志物的分类模型,例如随机森林模型、使用SIAMCAT的套索回归和XGBoost模型。每个案例都提供了详细的解释和可重现的R代码,使研究人员能够根据其特定的数据集调整示例。为了解决R包或其他工具频繁更新导致代码错误的问题,该项目将进行持续的更新和改进,和EasyAmplicon项目一样持续迭代。在此过程中,R包和其他工具将定期更新以保持兼容性并提高可用性,确保为用户提供可靠、最新的平台。此外,如果用户遇到代码错误,他们可以在GitHub上报告问题(https://github.com/YongxinLiu/MicrobiomeStatPlot/issues),我们将及时回复以解决问题。为了进一步提高可访问性,这些汇编分析通过“宏基因组”微信公众号共享,覆盖超过175,000名微生物组研究人员。截至2025年1月16日,这些共享案例已被浏览超过40,000次(表S1),使用户能够有效地构建和修改来自各种组学分析的组合图。
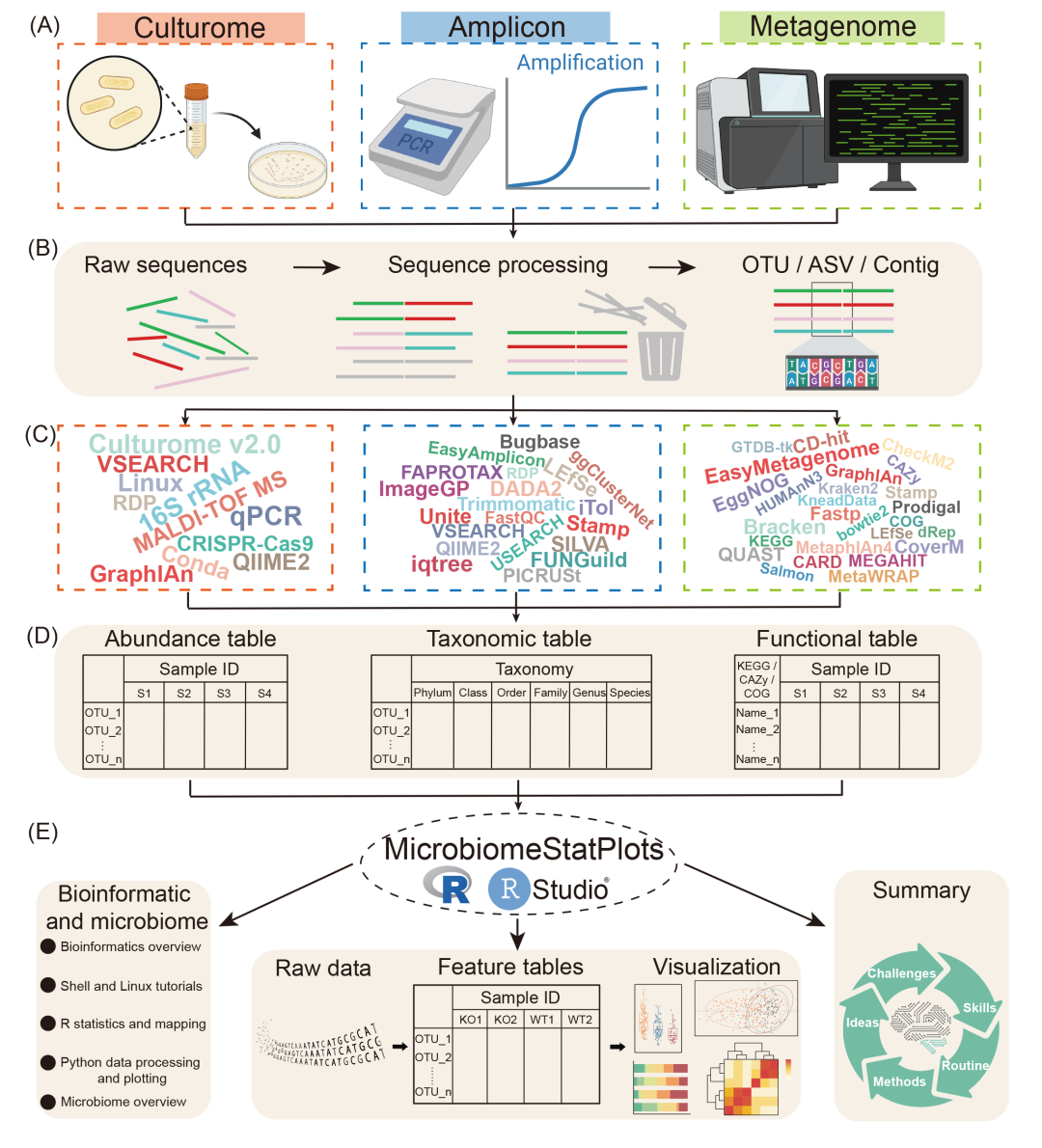
图2. MicrobiomeStatPlots的结构和组成
(A)MicrobiomeStatPlots涵盖多组学数据的分析,包括培养组、扩增子、宏基因组、病毒组和宏转录组等。(B)包括用于不同组学数据分析的生物信息学流程,有助于生成高质量的原始序列、序列处理和微生物组组成表。(C)该项目得到了各种不断更新的微生物组相关软件和多样化数据库的支持,确保了数据分析的稳健性和可靠性。(D)MicrobiomeStatPlots支持创建源自各种组学技术的物种组成和功能数据表,从而简化了微生物组数据处理。(E)该平台集成了基础生物信息学知识、多组学数据分析流程以及解释和可视化组学数据的方法。通过不断更新和迭代改进,MicrobiomeStatPlots旨在为推进微生物组研究提供全面支持。此图中的某些图形由BioRender(https://app.biorender.com/)创建。
案例1:培养组示例
MicrobiomeStatPlots提供了多种可视化选项来展示我们之前发布的培养组(Culturome)中微生物培养结果的更新(图3A-F)。箱线图说明了培养孔数与扩增子序列变体(ASVs)数量之间的关系,表明ASV数量随着孔数的增加而趋于稳定(图3A)。直方图显示了整个培养板上的微生物序列读长计数,虚线表示平均读长计数约为每板20,000次(图3B)。另一个直方图描述了所有培养孔中纯度百分比的分布,其中横轴代表不同的纯度,纵轴表示与不同纯度相对应的孔的比例;值得注意的是,超过80%的孔的纯度超过95%(图3C)。箱线图比较了培养中阳性对照和阴性对照的读长计数,显示阳性对照的读长计数明显更高(p < 0.001)(图3D)。使用ComplexHeatmap包绘制的热图显示了96孔板中每个孔的纯度,深红色和蓝色分别表示纯度较高和较低(图3E)。此外,系统发育树突出显示了培养微生物物种的组成,物种按其门着色,外圈表示培养细菌分离株的数量(图3F)。培养组单核苷酸多态性(SNPs)与丰度的关系和相互作用网络也用于进一步表征培养的微生物(图3G-H),如先前研究所述。彩色气泡图显示了培养物种的相对丰度与每种物种的全基因组单核苷酸多态性数量之间的关系,气泡大小代表分离基因组的数量,不同的颜色表示叶片SNPs(仅存在于一种基因型中)的比例。该图揭示了细菌相对丰度与SNP计数之间的正相关性(图3G)。相互作用网络通过igraph(https://github.com/igraph/igraph)和ggraph(https://github.com/thomasp85/ggraph)可视化了配对微生物属之间的正负相互作用。节点大小表示每个属中的分离株数量,而边表示相互作用类型,红色和蓝色分别代表正(促进生长)和负(抑制生长)相互作用。颜色深度表示一起培养的细菌菌落的面积(细菌菌落的形态特征之一)变化。颜色越深,面积随时间的变化越大。箭头表示相互作用的方向。例如,双歧杆菌促进粪杆菌属细菌的生长,但抑制瘤胃球菌的生长(图3H)。双歧杆菌属对粪杆菌属的影响是通过比较粪杆菌属菌落大小(以面积量化)与附近是否有双歧杆菌属菌落来测量的。使用Proksee创建的基因组环图显示了流感嗜血杆菌的基因组特征,注释了不同的类型和代表性基因(图3I)。
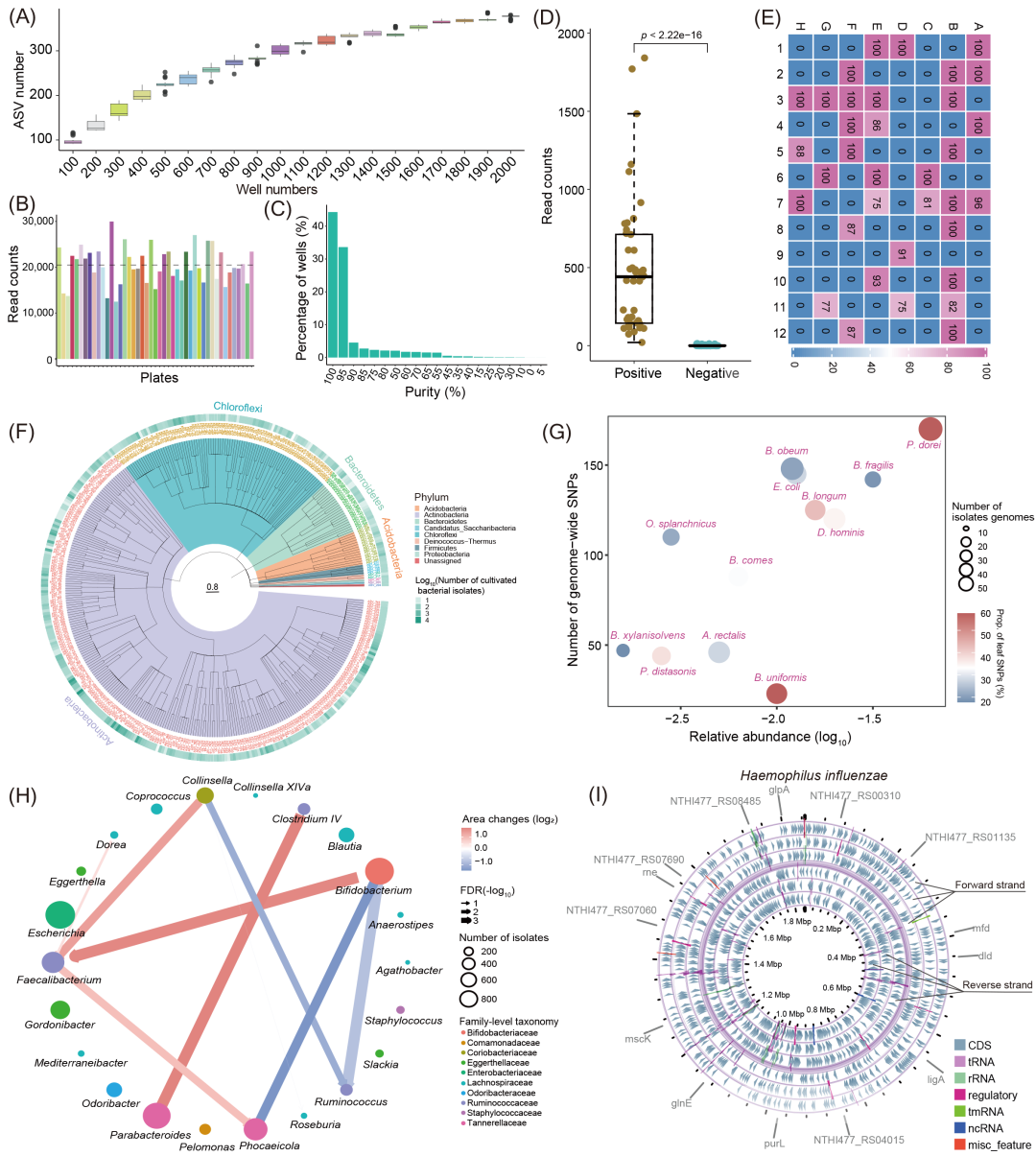
图3. MicrobiomeStatPlots中包含的培养组示例
(A)累积曲线显示了增加的孔数与培养的ASV数量之间的关系。(B)不同板的读长计数,平均每板20,000个。(C)不同孔的纯度分布。(D)阴性和阳性对照之间的读长计数比较。(E)96孔板中培养细菌的纯度百分比。(F)380个扩增子序列变体(ASVs)的系统发育树。分支颜色显示不同的细菌门。外圈显示每个分离的ASVs的相对丰度。(G)显著富集物种的相对丰度与全基因组单核苷酸多态性(SNPs)数量之间相关性的示例。(H)细菌属相互作用的网络可视化,其中红色边缘表示生长促进,蓝色边缘表示抑制。边宽表示相互作用显著性,而节点大小对应于分离株的数量,节点颜色反映科水平分类。(I)流感嗜血杆菌单个细菌基因组的示例。流感嗜血杆菌基因组的反向和正向链分别分为三个阅读框。三个内圈代表反向链,三个外圈代表正向链。反向链和正向链之间的圆圈代表基因组的主干。
案例2:扩增子示例
在本案例中,我们介绍了扩增子数据分析中常用的八种主要可视化形式,每种形式都提供了对微生物多样性、组成和关系的独特见解。α多样性衡量单个样本内的多样性(包括丰富度和均匀度),通常使用箱线图来比较组间的数值。α多样性的差异可以使用方差分析(ANOVA)进行统计评估(图4A)。β多样性评估样本间分类群组成和群落结构的差异,通常通过主坐标分析(PCoA)在散点图中可视化。可以使用R中的vegan包(https://github.com/vegandevs/vegan)绘制,可以使用vegan包中的“adonis”函数进行置换多元方差分析(PERMANOVA)来评估组间β多样性的差异(图4B)。维恩图主要用于评估不同组间共享和独特元素的定量关系。可以使用R软件中的ggVennDiagram包绘制这些图。还建议使用EVenn的网络版本(http://www.ehbio.com/test/venn/#/)进行绘图(图4C)。堆叠柱状图提供了组间微生物组成变化的整体可视化,并可以在不同分类水平上展示物种组成。可以使用R中的ggplot2包生成这些图(图4D)。显著性相关热图是一种统计工具,用于评估变量之间关系的强度。相关系数显示为热图,颜色强度反映变量关联的强度和接近度。这些图的生成由R中的ggcor或linkET(https://github.com/Hy4m/linkET)包实现(图4E)。带标签的火山图是一种常用于显示显著上调和下调的基因或物种的常用方法,从而帮助研究人员快速识别数据中的显著差异(图4F)。STAMP图经常用于展示组间物种或功能的差异分析,通过条形图、置信区间分布和p值展示成对比较(图4G)。系统发育树对于可视化物种间的进化关系是必不可少的。这些可以使用R中的ggtree包生成(图4H)。

图4. MicrobiomeStatPlots中包含的扩增子示例
(A)比较三个组(AT、AO和AP)之间的操作分类单元(OTU) 丰富度。(B)使用主坐标分析(PCoA)进行的Beta多样性分析显示组(BS、R和RS)之间的微生物群落结构差异。(C)维恩图显示三个区域(Root、Rhso和Tbuso)之间相同或不同的共享和唯一OTU的数量。(D)堆叠柱状图显示了门级别上不同组(AT、AO、AP和AK)的细菌组成。(E)Mantel检验热图显示了土壤特性与细菌种类和代谢之间的Pearson相关性。(F)火山图显示不同组之间显著富集(红色)或耗尽(蓝色)的属。(G)STAMP图显示了患者组和健康组之间的功能通路差异。左侧面板显示患者组(绿色条)或健康组(橙色条)的相对丰度。右图显示差异的p值和95%置信区间。(H)不同扩增子序列变体(ASVs)之间的系统发育关系,分支按不同科水平着色。
案例3:宏基因组示例
对于宏基因组数据,MicrobiomeStatPlots提供了不同方面的综合可视化解决方案(图5)。一方面,几种基本绘图类型(如“基本柱状图”、“折线图”和“散点图”)是基础选项。例如,Upset图可以显示组间的交叉物种或基因家族,突出显示其独特和共享的组成部分(图5A)。另一方面,高级可视化将分析方法集成到绘图过程中。例如,共丰度网络说明了物种与KO基因之间的相关性,其中SparCC方法集成到结果生成过程中(图5B)。系统发育树圆形图可视化了不同物种间的系统发育关系,并比较了各物种在各组间的相对丰度,生成系统发育树圆形图的代码已集成到EasyMetagenome流程(https://github.com/YongxinLiu/EasyMetagenome),使用“gtdbtk”方法生成无根树(图5C)。热图和直方图的组合显示了组间的差异分析结果以及每个物种在其对应组中的相对丰度,热图上的星号表示组间显著差异(图5D),差异分析方法MaAsLin2已集成到绘图过程中。此外,我们还将多种宏基因组数据分析方法集成到MicrobiomeStatPlots中。例如,anpan方法(由bioBakery项目开发)(https://github.com/biobakery/anpan)可用于微生物群菌株分析。MicrobiomeStatPlots提供了在R中使用anpan的指南,解决了操作错误,并支持跨组比较Alistipes shahii等菌株的基因组成(图5E)。此外,使用anvi’o进行泛基因组分析可以在物种或属水平进行基因组探索。在这里,我们以Alistipes shahii为例,展示其目前已知的部分基因组信息(图5F)。MicrobiomeStatPlots中包含泛基因组分析的分步教程,扩展了其在宏基因组研究中的实用性。
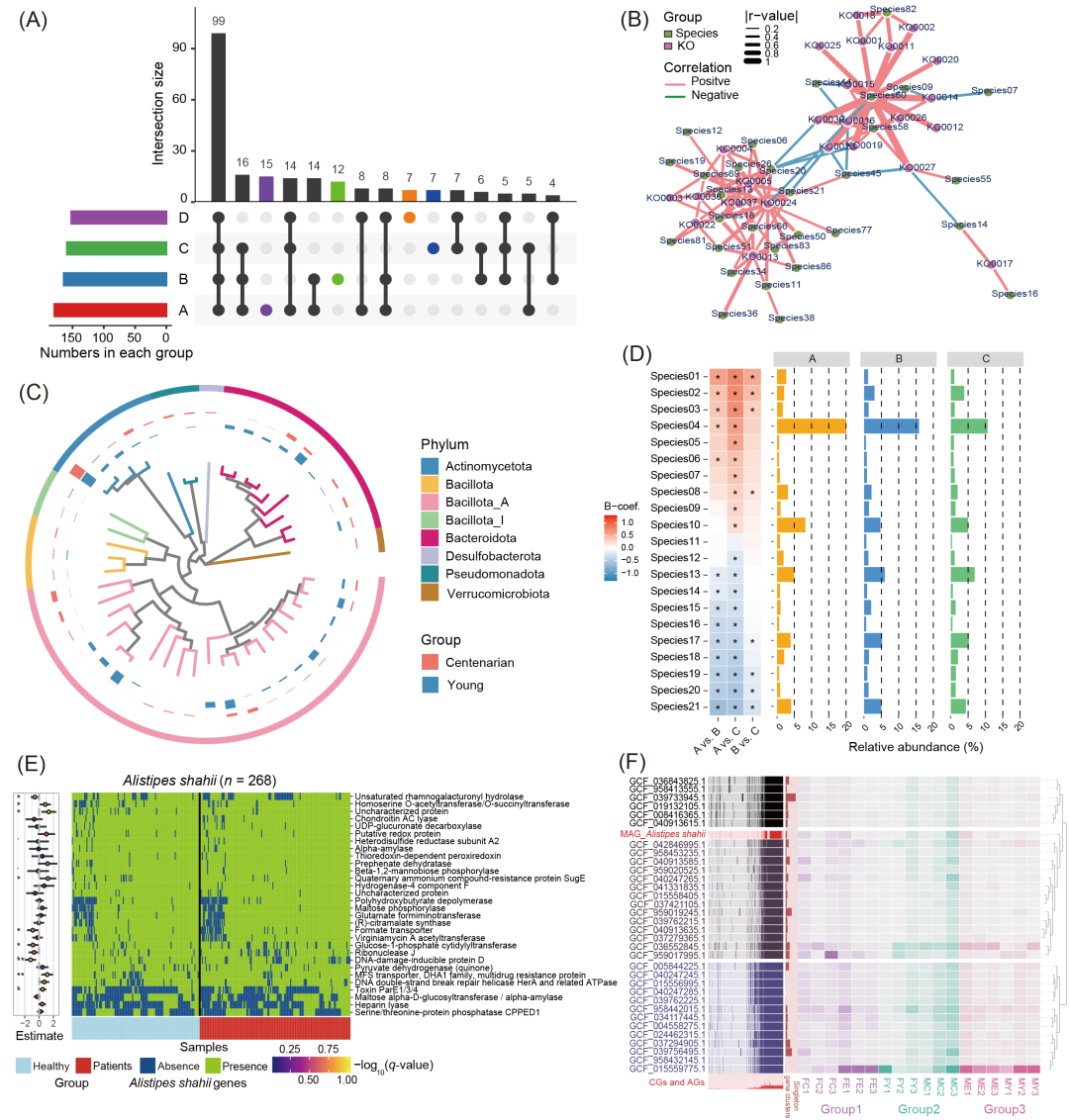
图5. MicrobiomeStatPlots中包含的宏基因组示例
(A)Upset图显示了各组间共享和独特物种的数量。(B)共丰度网络显示了物种与KO基因之间的正相关和负相关。使用组成数据稀疏相关性(SparCC)分析方法计算每对的相关性和p值。(C)系统发育树显示了使用人类宏基因组序列数据注释的不同宏基因组组装基因组(MAG)的系统发育关系。不同的门级别用不同的颜色标记。分支树外的两圈直方图分别显示了年轻人和百岁老人组中每种MAG物种的流行率。(D)宏基因组物种的差异分析。左侧热图显示了组间显著性以及富集(红色)或下调(蓝色)趋势。右侧的三个组合条形图显示了每组中每种物种的相对丰度。(E)热图显示了与疾病发生相关的Alistipes shahii基因,显示了使用anpan进行微生物群菌株分析的结果。颜色显示在宏基因组检测到的Alistipes shahii菌株中UniRef90基因家族的缺失(蓝色)或存在(绿色)。横轴代表样本。纵轴显示与UniRef90基因家族相对应的功能途径。回归统计的系数估计值(左侧的点和误差线)显示了每个基因对结果(健康或患者)的影响。不同颜色的点代表图例中显示的不同q值。显著影响(*代表p < 0.05,**代表p < 0.01)用星号标记。(F)使用宏基因组序列数据对Alistipes shahii基因组进行泛基因组分析。左侧的热图显示每个基因组中检测到的(暗区)或未检测到的(亮区)基因。左下方柱状图描述了核心基因组(CGs)/附属基因组(AGs)的比例,红色代表CGs的比例。中间的水平条形图显示了每个基因组中单个基因簇的数量。右侧渐变色热图描绘了不同样本中基因组的中位覆盖率(第1组为紫色,第2组为绿色,第3组为红色)。所有基因簇按Alistipes shahii基因组中的基因簇频率排序(右侧的簇树)。
MicrobiomeStatPlots的未来发展方向
MicrobiomeStatPlots是一个用于多组学数据可视化和解释的综合项目。虽然目前的重点主要放在培养组、扩增子和宏基因组上,但将范围扩大到其他组学数据的需求日益增长。MicrobiomeStatPlots的当前版本包含有限的代谢组学、宏蛋白质组学、病毒组学和宏转录组学示例和资源。例如,使用水平分面堆积图比较宏基因组和宏转录组之间的代谢结果,展示组成百分比差异(图6A)。热图突出显示了健康组和患者组之间的代谢物含量差异,清楚地展示了哪些代谢物发生了剧烈变化(图6B)。棒棒糖图显示了各组间每种代谢物的相对重要性,提供了各组特异性代谢物的直观表示(图6C)。Spearman相关网络揭示了物种与代谢物之间相关性的方向(正相关或负相关)和强度(用连线宽度表示)(图6D)。可以使用STRING(https://string-db.org/)等在线平台生成蛋白质-蛋白质相互作用网络,从而深入了解蛋白质簇和相互作用类型(图6E)。系统发育树显示了不同组中的病毒操作分类单元(vOTUs)及其各自的细菌宿主。外圈的条形图显示了每个vOTU在每个组内的流行率。该图可以清楚地显示不同组之间vOTUs的组成差异(图6F)。尽管取得了这些进展,但该项目仍需要进行大规模扩展才能完全涵盖其他组学数据。提出了以下未来发展方向:(1)开发和整合用户友好的代谢组学、宏蛋白质组学、病毒组学和宏转录组学流程,以拓宽MicrobiomeStatPlots的范围。(2)编译更多不同组学类型的数据分析和可视化示例,为解释和绘图提供全面的资源。(3)将绘图代码直接集成到组学数据分析流程中,实现从特征表生成到可视化和解释的无缝过渡。对于不同的分析流程,首先必须对不同组学中常用的数据可视化和解释案例进行分类。然后,应将代表性的分析方法和可视化样式集成到适当的分析流程中,并为每种类型的分析和可视化提供专用的模块(例如,物种组成概述、差异分析和模型分析)。(4)鼓励微生物学研究人员为项目内数据分析、可视化和解释工具的持续改进做出贡献。参与度的提高将增强项目的可用性和影响力。通过解决这些方向,MicrobiomeStatPlots旨在成为一种更加通用和全面的微生物组数据分析工具,使组学研究更加易于获取和高效。
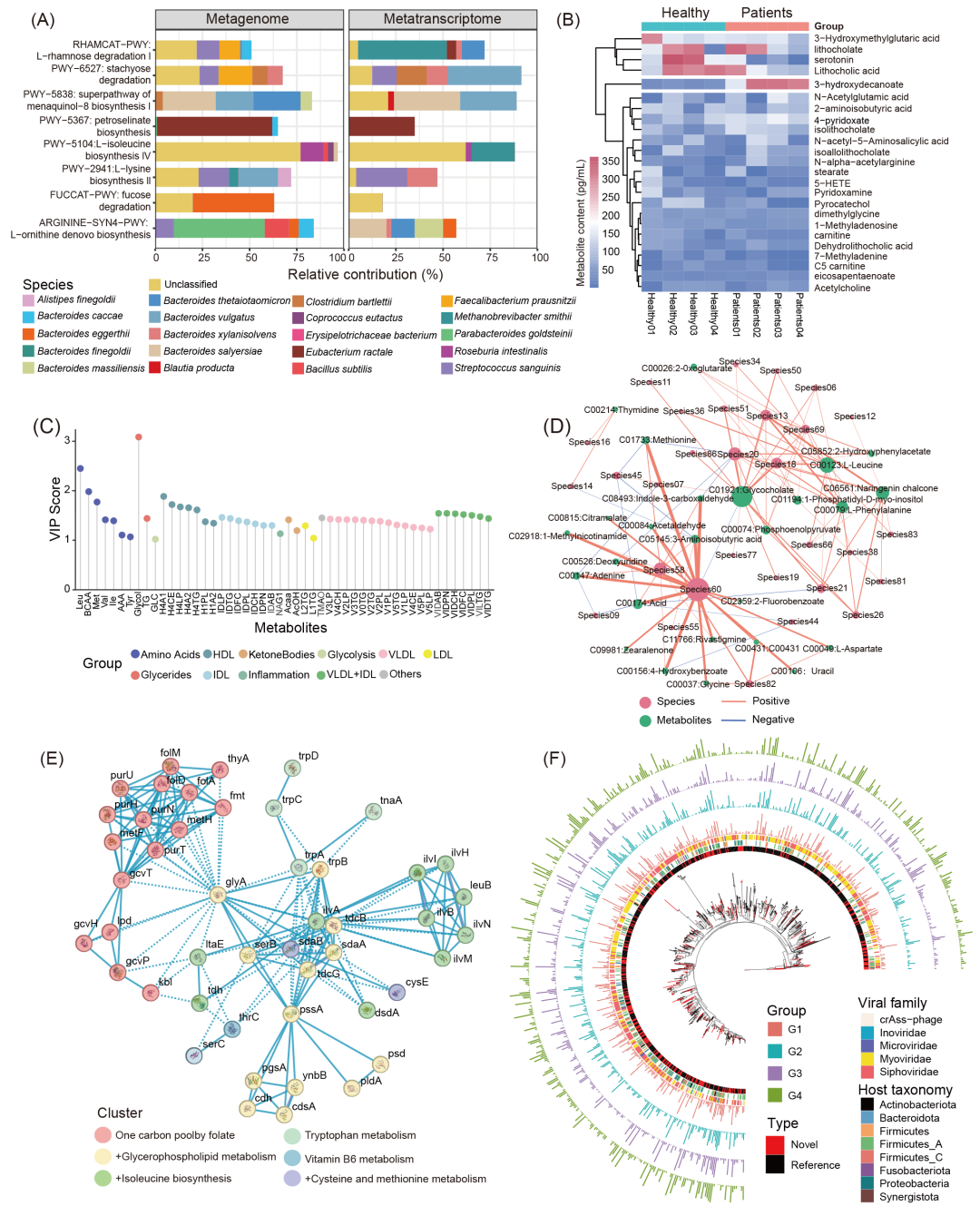
图6. MicrobiomeStatPlots的未来发展方向
(A)宏基因组与宏转录组代谢结果对比示例。堆叠图显示了各物种对各功能通路的相对贡献百分比。(B)健康组与患者组代谢物含量对比热图。(C)变量对投影(VIP)分数的影响表示每种代谢物的重要性。不同的颜色代表不同的代谢物类别。(D)物种与代谢物的斯皮尔曼相关网络示例。橙色和蓝色分别表示正相关和负相关。连线宽表示相关强度。(E)蛋白质-蛋白质相互作用网络示例,并按簇分组。(F)不同病毒操作分类单元(vOTUs)和不同组(G1、G2、G3和G4)的附加特征的系统发育树示例。树上的红色分支代表以前未描述过的(新型)vOTU。最内圈的圆圈显示了新型(红色)和参考(以前描述过的)vOTUs的区别。向外的第二个圆圈代表不同vOTUs的不同宿主,并根据细菌门级别着色。向外的第三个彩色圆圈显示了不同科水平的vOTU。由条形图组成的四个最外圈代表四个组中每个vOTU的流行率(G1为红色,G2为青色,G3为紫色,G4为绿色)。
讨 论
MicrobiomeStatPlots为微生物组数据分析、绘图和解释提供了一种可重复且用户友好的解决方案,为多组学数据的可视化和理解做出了重要贡献。尽管已有多个分析平台用于微生物组数据分析和可视化,如用于宏基因组数据分析的MicrobiomeAnalyst、用于代谢组学数据分析的MetaboAnalystR和用于原核生物基因组分析的IPGA平台,但这些平台通常侧重于组学数据的特定方面。其他几个数据分析和可视化平台在组学研究中被广泛使用。例如,Sangerbox平台为临床生物信息学提供了用户友好的工具,而Majorbio Cloud提供了多组学数据分析和可视化。ImageGP是微生物组数据分析的流行工具,而Wekemo Bioincloed提供了22种工作流程和65种可视化工具。然而,这些平台仍然存在局限性,例如某些平台的某些功能需要付费才能使用,解释支持有限,缺乏可定制、可重现的代码。MicrobiomeStatPlots通过提供80多个多组学数据可视化和解释案例来弥补这些不足,这些案例涵盖培养组、扩增子测序、宏基因组测序以及代谢组学、宏转录组学、宏蛋白质组学和病毒组学的初始示例。该项目不仅提供可视化,还集成了详细的案例解释、可重现的代码和可访问的示例数据集,使用户可以调整和扩展分析。
此外,MicorbiomeStatPlots的一大特色是其对绘图方案和示例的详细解释。这种方法通过提供全面的图形再现和结果解释指导,弥补了现有平台的不足。用户可以访问原始代码和示例数据表,从而促进可视化的可重复性和扩展性。这些功能解决了常见的痛点,例如缺乏绘图数据和数据格式不一致,这些都阻碍了图形的快速再现。该项目还包括R软件和Linux系统的基本教程,为不熟悉这些工具的用户解决障碍。这个知识库确保微生物组数据的分析、绘图和解释更容易被更广泛的受众所接受。MicrobiomeStatPlots在组学数据的全面覆盖、绘图数据和代码的可用性以及可视化的呈现和解释方面,在其他已发布的在线平台中脱颖而出。它在这些领域具有显著优势,使其成为现有工具的宝贵补充。
尽管本研究提供了大量微生物组可视化和解释示例,但仍存在一些局限性。一个局限是缺乏用户友好的图形界面或用于调整可视化的交互式工具。这对于不熟悉编程或希望更直观地根据特定研究需求定制图表的用户带来挑战。将来,随着我们积累更多用户友好的可视化案例,可能需要一个更简单、更易于访问的交互式界面来帮助没有编程经验的用户。或者,这些现有的可视化案例可以集成到交互式平台中,例如Wekemo Bioincloud和ImageGP。此外,虽然我们为已发表的论文中的大多数可视化和示例提供了简短的介绍和初步解释,但解释的深度仍然有限。在未来的更新中,我们旨在提供更全面的分析,解释各种图形在微生物组研究中的作用,并指导用户如何解释这些图形中的元素。我们还计划对分析代码中的每一步提供更详细的解释。此外,尽管EasyAmplicon和EasyMetagenome等流程中存在一些集成的可视化和解释工具,但仍然缺乏将这些可视化工具无缝整合到更广泛的研究流程中的详细综合框架。未来,我们计划整理和分类当前的微生物组分析方法和可视化案例,将更多易用的工具纳入现有的数据分析工作流程中。为了扩大国际用户群,MicrobiomeStatPlots为每个可视化和解释案例以及GitHub上的用户指南(https://github.com/YongxinLiu/MicrobiomeStatPlot)提供了中英文双语描述。然而,许多内容仍然是用中文编写的。为了进一步扩大MicrobiomeStatPlots的影响力,我们将致力于翻译和分享这些中文材料,以帮助提高该平台的国际知名度和用户参与度。
展望未来,MicrobiomeStatPlots将继续发展,纳入更多代谢组学、宏转录组学、宏蛋白质组学和病毒组学数据案例。培养组、扩增子和宏基因组的分析、可视化和解释也将进一步扩展。此外,将代码案例集成到现有流程中,例如培养组(Culturome)、易扩增子(EasyAmplicon)和易宏基因组(EasyMetagenome)(https://github.com/YongxinLiu/EasyMetagenome)流程,将使用户能够直接从原始数据表中提取数据,进行无缝绘图。增加社区参与和合作将进一步巩固MicrobiomeStatPlots作为一个全面且适应性强的微生物组研究资源的地位。
结 论
总之,MicrobiomeStatPlots为快速增长的多组学数据集的可视化和解释提供了有价值的解决方案,尤其是对于培养组、扩增子和宏基因组数据。该项目的未来迭代旨在为其他组学数据(如代谢组学、宏蛋白质组学、病毒组学和宏转录组学)提供更多资源。该集成项目结合了三种广泛使用的组学数据分析流程(培养组、扩增子和宏基因组)、80多个用于分析和解释多组学数据的示例以及更新的微生物组数据分析最佳实践流程。通过提供可重复的数据分析和可参考的结果解释,MicrobiomeStatPlots项目可以为微生物组数据分析和解释提供个性化的解决方案。
方 法
MicrobiomeStatPlots被设计为一个基于代码的框架,在Linux系统上使用R软件和Shell脚本运行。对于多组学数据分析,使用了专门针对微生物组研究定制的各种软件工具和平台。由于必须为每种组学类型获取特征表,因此在下游数据处理和解释之前执行上游数据分析流程。培养组、扩增子和宏基因组数据分析的主要步骤可概括如下:对于培养组分析,关键步骤包括准备映射文件、合并双端序列、样本分割、测序深度计算、引物切除、ASV构建、物种注释和系统发育树生成。扩增子测序分析包括文件准备、读取合并、引物修剪、OTU或ASV聚类、特征表创建、多样性计算和物种注释。宏基因组分析包括数据预处理、基于读长和基于组装的分析、样本分箱和细菌基因组分析(https://github.com/YongxinLiu/EasyMetagenome)。
使用了大量R包来处理特征表、绘图和结果解释。表S2提供了用于每个分析或绘图示例的软件包的完整列表。截至目前,MicrobiomeStatPlots的可视化和解释部分已经使用了190多个R包。绘图结果主要参考已发表论文中的例子进行解释。每个案例包含几个部分:(1)分析或图表的简要介绍,包括其组成元素的解释及其在表示微生物组数据中的作用。(2)至少提供一个已经发表文献的例子,并详细解释了图例和结果。(3)提供可重复的分析和绘图代码以及示例数据,以确保代码的无缝运行。原始代码和示例数据集的加入使用户能够调整其数据,从而绘制一致且可重复的图表。为了帮助R或Linux经验有限的用户,“生物信息学和微生物组”部分包含了有关使用R软件和Linux系统基础知识的教程,确保微生物组数据分析、绘图和解释的可访问性和广泛可用性。
代码和数据可用性
MicrobiomeStatPlots是免费提供的,以Shell和R实现,易于下载和使用。该项目在GitHub上提供https://github.com/YongxinLiu/MicrobiomeStatPlot。所有图形数据都可以通过GitHub或补充表格访问。目前使用的所有R包都可以通过https://pan.baidu.com/s/1wKSPuOZueJ0H_EsNsnjBYg?pwd=ze7q访问。补充材料(表格、图形摘要、幻灯片、视频、中文翻译版和更新材料)可在网上DOI或iMeta Science中找到http://www.imeta.science/。
引文格式:
Defeng Bai, Chuang Ma, Jiani Xun, Hao Luo, Haifei Yang, Hujie Lyu, et al. 2025. "MicrobiomeStatPlots: Microbiome statistics plotting gallery for meta-omics and bioinformatics." iMeta 4: e70002. https://doi.org/10.1002/imt2.70002
作者简介

白德凤(第一作者)
● 中国农业科学院深圳农业基因组所博士后。
● 博士毕业于中国科学院动物研究所。目前研究方向为宏基因组数据分析及挖掘,肠道微生物和疾病关系研究。相关学术成果已发表于iMeta, Landscape Ecology, Zoological Research等期刊。

马闯(第一作者)
● 中国农业科学院深圳农业基因组所客座硕士,安徽农业大学园艺学院研三在读。
● 主要研究方向为百岁老人肠道病毒组、土壤微生物组。

荀佳妮(第一作者)
● 中国农业科学院深圳农业基因组所生物信息学硕士在读,本科毕业于华北理工大学生命科学学院。
● 主要研究方向为宏基因组数据分析和挖掘。

王瑶(通讯作者)
● 中国农业科学院深圳农业基因组所博士后。
● 博士毕业于德国哥廷根大学。研究方向为植物-微生物互作,相关学术成果已发表于PLoS Pathogens、Frontiers in Microbiology等期刊。

刘永鑫(通讯作者)
● 中国农业科学院深圳农业基因组研究所研究员,微生物组与营养健康团队首席,博士生导师。iMeta期刊执行主编,宏基因组公众号创始人,中国微生物学会微生物组专委会委员。
● 研究方向聚焦微生物组学方法开发、功能挖掘和科学传播,以第一或通讯作者(含共同) 在Nature Biotechnology、Nature Microbiology 等发表研究论文40余篇,在Current Opinion in Microbiology、Protein Cell等撰写综述20余篇,合作在Science、Cell Host & Microbe等期刊发表论文20余篇,累计发文80余篇,被引用23000余次,连续入选全球前2%顶尖科学家榜单。主持国自然青年/面上,中国科学院/中国农科院/深圳市等人才项目。创办宏基因组公众号,17万+同行关注,累计阅读量超6千万。主编《微生物组实验手册》专著,联合152家单位的352位同行参与,共同打造本领域长期更新的中文百科全书。发起iMeta期刊并任执行主编,影响因子23.8位列微生物学科研究类期刊全球第一。为Cell Host & Microbe、Nature Communications、Microbiome、ISME、NAR等期刊审稿300余次。
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

1卷1期

1卷2期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

魔乐社区(Modelers.cn) 是一个中立、公益的人工智能社区,提供人工智能工具、模型、数据的托管、展示与应用协同服务,为人工智能开发及爱好者搭建开放的学习交流平台。社区通过理事会方式运作,由全产业链共同建设、共同运营、共同享有,推动国产AI生态繁荣发展。
更多推荐


 0
0 0
0- 0



 已为社区贡献33条内容
已为社区贡献33条内容

所有评论(0)